






















テクニカルインフォメーション
アガロースゲル電気泳動 基本操作
アガロースゲルを使用した電気泳動は、核酸を分析する手段として一般に広く使われている方法です。ゲルを緩衝液に沈めて電気泳動をすることから、海中の潜水艦(サブマリン) に例えて、この方法はサブマリン電気泳動といわれています。
DNAを構成しているヌクレオチドはリン酸残基によりマイナスに荷電しています。アガロースゲルにDNA(試料)を添加し、緩衝液中で電気を流すと、DNAはプラス側(陽極)に移動します。この時、アガロースゲルの網目構造により大きなサイズのDNAはゆっくりと動くのに対して、小さいDNA はより速く動きます。この原理を利用してDNAを分離する方法がアガロースゲル電気泳動です。
タンパク質の電気泳動でよく使用されるポリアクリルアミドゲルも同じような網目構造を持ちますが、アガロースゲルの方がその網目が大きく、DNAの大きさによって使い分けをします。アガロースゲルは、約0.5~20kbpのDNAを分離するのに適しているといわれています。
今回は、アトーの製品を使用したサブマリン方式のアガロースゲル電気泳動の基本操作に関してご紹介します。
実験の流れ
1.サンプル調製
細胞や組織、血液など様々な試料からDNAを抽出します。抽出したDNAをEzApplyDNAと混合し、泳動サンプルを調製します。
使用する製品
ローディングバッファー
(EzApplyDNA)

4.ゲルの染色と撮影
泳動後のゲルをEzFluoroStain DNAやEzPreStain DNA&RNAまたはエチジウムブロマイド(EtBr)で染色します。染色後のゲルを蛍光撮影し、分離したバンドを確認、解析します。
使用する製品
ゲル染色液
(EzFluoroStainDNA, EzPreStain DNA&RNA)
実験の前に
DNA、RNAを切断する酵素であるDNase(デオキシリボヌクレアーゼ ,Deoxyribonuclease)やRNase(リボヌクレアーゼ,Ribonuclease)は、自然界のありとあらゆる所に存在します。
実験に用いる試薬や器具にヌクレアーゼがコンタミすると、サンプルであるDNAやRNAが分解され、実験の失敗に繋がります。
DNAやRNAを用いる実験を行う際はDNaseやRNaseに十分な注意が必要です。
DNAを取り扱う上での注意
DNAを分解する酵素であるDNaseは、普段の我々の手にたっぷりと存在します。DNAを用いる実験を行う際は、必ずゴム手袋を着用し、適宜エタノールを使用します。
また、DNaseはオートクレーブの滅菌処理で不活性化することができます。
なので、実験で使用するチップやチューブなどのプラスチック製品、サンプル調製や電気泳動で使用するバッファー類や蒸留水についても、必ずオートクレーブ処理を行ってから実験に使用します。
ただし、電気泳動後にDNAのバンドを回収しない場合は、ゲルや泳動バッファーの滅菌は不要です。
RNAを取り扱う上での注意
RNAはとても敏感な核酸であり、RNaseによって迅速に分解されてしまうため、RNAを使用する実験はRNaseフリーの条件下で行うことが絶対です。
RNAを分解する酵素であるRNaseは、失活しにくい上に、オートクレーブや紫外線(UV)照射、エタノールに対しても極めて高い耐性があるといわれています。
実験の際に手にゴム手袋を着用することは絶対条件ですが、それ以外にも蒸留水やバッファーをDEPC処理しておく必要があります。
DEPCとは…
ジエチルピロカーボネート(Diethylpyrocarbonate,DEPC)はRNase を失活させるための試薬です。
DEPCは非常に強力なRNase 阻害効果を持っており、RNA を扱う際に分解のリスクを避けるためによく用いられています。
しかし危険物指定されている試薬であり、発がん性、引火性があるため、取り扱いには十分に注意し、作業はゴム手袋を必ず着用し、ドラフト内で行います。
DEPC水の作り方
1.密閉できる容器に滅菌済みの超純水を1000mL入れ、DEPCを1mL添加する(最終濃度0.1%になるようにDEPCを添加します)。
2.容器をよく振り、DEPCを溶解する。
3.37℃で2時間以上置いておく(室温でオーバーナイトでも可)。
4.容器のふたを緩め、オートクレーブ(121℃、20分)をかけてDEPCを不活性化する。
5.オートクレーブ後、室温で保存する。
RNAの実験に用いる水やバッファー類には、必ずDEPC水を使用します。
電気泳動で用いるゲルを作製するためのバッファーも必ずDEPC水で調製したものを使用します。
プラスチック製品や電気泳動槽も、RNaseフリー処理が必要となります。
これらは、きれいに洗浄し、95%エタノールで軽くすすいでおきます。
3%の過酸化水素水(超純水で作製したもの)で浸漬し、室温で15分ほど置いておきます。
その後DEPC水で3回程度すすぎ、乾かすと器具のRNaseフリー処理ができます。
実験方法
サンプル調製
1.細胞や組織などの試料からDNA抽出キットを用いて、DNAを抽出します。
2.EzApplyDNA (6×濃度)1µLに、抽出したサンプル5μLを加えて混合します。
【パラフィルムを使用して試料調製する場合】
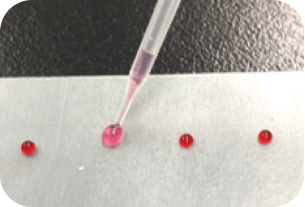
パラフィルム上にEzApplyDNAを1µL滴下し、そこに抽出したサンプル5μLを加え、ピペッティングで混合します(左図参照)。
ゲルの作製
サブマージ ミニでは、ゲルトレイ(L)1枚あたり25~40mL、(S)1枚あたり15~20mLのアガロースゲル溶液が必要です。
アガロースゲル溶液量はゲルの厚さにより異なり、最適なゲル厚は4~6mmです。
ゲルが厚いとバンドがぼやけて見えたり、バックグラウンドが高くなる原因にもなります。
1.分離するDNAの大きさに応じて、アガロースを目的濃度になるように秤量します(下表参照)。
| アガロースゲル濃度(w/v) | 分離するDNAのサイズ(bp) |
|---|---|
| 0.6% | 1,000~20,000 |
| 0.7% | 800~10,000 |
| 1.0% | 500~7,000 |
| 1.2% | 400~6,000 |
| 1.5% | 200~3,000 |
| 2.0% | 100~2,000 |
ゲルバッファー量(mL)×ゲル濃度(%)=アガロース量(g)
例えば…1.0%のLサイズのアガロースゲルを作製したい場合
40mL(ゲルバッファー量)×1.0%(ゲル濃度)=0.4g(アガロース量)
0.4gのアガロースを40mLのゲルバッファーに溶解し、ゲルを作製する。
2.1.にゲルバッファー(1×EzRunTAEまたは1×EzRunTBE)を必要量添加します。
3.電子レンジまたは湯せんで加熱し、アガロースを完全に溶解します。
※溶解後のアガロースゲル溶液は突沸する可能性があり、非常に高温のため、火傷をする恐れがあります。必ず耐熱手袋をして作業してください。
※加熱中のアガロースゲル溶液の突沸を防止するために、溶液量の2~5倍の三角フラスコやビーカーを使用することをお勧めします。
4.アガロースゲル溶液を約60℃くらいまで冷まします。
※高温のアガロースゲル溶液をゲルトレイに流しこむと、ゲルトレイが変形することがありますので、必ず冷ましてください。
5.適温に下がったアガロースゲル溶液をゲルトレイに流します。
※ゲル中に空気が入らないようにゆっくりと静かに注ぎいれます。
※空気が入ってしまった場合は、チップの先などでつついて、空気を除去します。
6.ゲル溶液が固まる前に、ゲルトレイにコウムをセットします。
7.ゴミが入らないようにラップをかけ、室温で30~60分静置してゲルを固めます。作製したゲルは1×EzRunTAEまたは1×EzRunTBEバッファー中で約1週間、冷蔵保存が可能です。
※EzPreStainDNA&RNAはゲル溶液、泳動bufferに添加による先染めが可能です。詳細は製品取扱説明書を参照ください。
電気泳動
1.作製したアガロースゲルをゲルトレイごと電気泳動槽(サブマージ ミニ、電源搭載型電気泳動装置)に入れます。
※ゲルトレイについた余分なゲルは取り除いておきます。
2.電気泳動槽に泳動バッファー(ゲル作製に使用したものと同じバッファー)を200~230mL入れます。
3.調製したサンプル溶液を空気が入らないように、ゆっくりアプライします。
4.上部カバーをセットします。
5.下表を参照し、泳動条件を設定して、スタートボタンを押して電気泳動を開始します。
| 設定電圧 | 設定時間 |
|---|---|
|
50V |
60min |
| 100V | 30min |
| 150V | 20min |
ワンポイントアドバイス【泳動バッファーの量はゲルの上面より1~3mm上までいれる】
泳動バッファーの量が多すぎると、ゲル上面部位のバッファー中を流れる電気が多くなるため、泳動時間が長くなります。
また、泳動パターンの乱れの原因にもなりますので、バッファー量の入れすぎには注意が必要です。
ゲルの染色と撮影
1.50mLの1×EzRunTAEまたは1×EzRunTAEにEzFluoroStainDNAまたはEzPreStainDNA&RNAを5µL添加し、混合します。
※EzFluoroStainDNAやEzPreStainDNA&RNAが完全に溶解せず、不溶物が見られることがあります。
その場合は、まず蒸留水500µLでEzFluoroStainDNAまたはEzPreStainDNA&RNAを5µLを希釈し、その後1×EzRunTAEまたは1×EzRunTBEを添加し、50mLの染色液を作製します。
2.上部カバーを外し、ゲルをゲルトレイごと取り出します。
※電気泳動直後は、泳動バッファーが高温になることがあります。火傷に注意してください。
3.樹脂製のタッパーに染色液を入れ、その中に泳動後のゲルを入れて、浸漬します。この時ゲルトレイは外し、ゲルのみを染色液に入れます。
※必ず樹脂製のタッパーを使用します。ガラス容器を使用すると、蛍光試薬がガラスに吸着します。
4.タッパーをアルミホイルなどで遮光し、室温で10~30分間インキュベーションします。
5.インキュベーション後、ヘラなどを使用して、ゲルを慎重に取り出します。
※脱色操作は不要です。染色後の染色液は4℃で約1週間、遮光保存が可能です。
6.取り出したゲルは、撮影用トレイまたはラップの上にのせます。
7.ゲル撮影装置にゲルをセットし、Cyan LED またはUV光源で励起し、撮影します(下表参照)。
| UV励起 | 260~370nm | フィルター:500~580LP |
|---|---|---|
| Cyan LED励起 | 440~500nm | フィルター:500~580LP |
自作の場合
下表は、アガロースゲル電気泳動で使用する主な試薬の組成です。
より再現性良く、簡便に実験を行う際は、ぜひアトーの試薬をご利用ください。
| 試薬名 | 組成 |
|---|---|
| ローディングバッファ(5×Conc.) | 50% Glycerol,1mM EDTA(pH8.0)、0.25% Bromophenol Blue0.25%、Xylene Cyanol FF |
| TAE バッファー(50×Conc.) | 2M トリス、1M 酢酸、50mM EDTA |
| TBE バッファー(10×Conc.) | 500mM トリス、485mM ホウ酸、20mM EDTA |
| 染色液 | 10mg/mL エチジウムブロマイド、1×TAEまたは1×TBEバッファー |
エチジウムブロマイド(EtBr)を染色に使用する場合は、以下のような手順で行います。
エチジウムブロマイドは発がん性物質ですので、人体に接触しないように、取り扱いには十分気を付けてください。
1.50mLの1×EzRunTAEまたは1×EzRunTBEに50µLのエチジウムブロマイド(10mg/mL)を添加し、混合します。
2.染色液にゲルを浸漬し、アルミホイル等で遮光しながら5~30分間、染色します。
3.染色後、へら等を使用して、ゲルを慎重に取り出します。バックグラウンドが高い時は、蒸留水に浸し、30~60分脱色します。
4.ゲル撮影装置にゲルをセットし、UV照射で撮影します。
実験例/豆知識
実験例:泳動バッファーの違い
アガロースゲル電気泳動で使用するバッファーは、TAE(Tris-Acetate-EDTA)バッファーとTBE(Tris-Borate-EDTA)バッファーの2種類が主流です。
10kbp以上の大きなDNAを分離したい場合は、TAEバッファーを使用します。TAEは緩衝能が低いため、短時間の電気泳動に適しています。
また、直鎖状二本鎖DNAはTBE中よりもTAE中の方が10%ほど早く移動するといわれています。価格はTBEよりも圧倒的に安価です。
1kbp以下の小さなDNAで、泳動後にDNAを抽出しない、または回収率を気にしない場合には、TBEバッファーを使用します。
TBEは緩衝能が高く、移動度が小さいので、小さいフラグメント(<1kbp)に対してより高い解像度を得ることができ、バンドが鮮明になります。
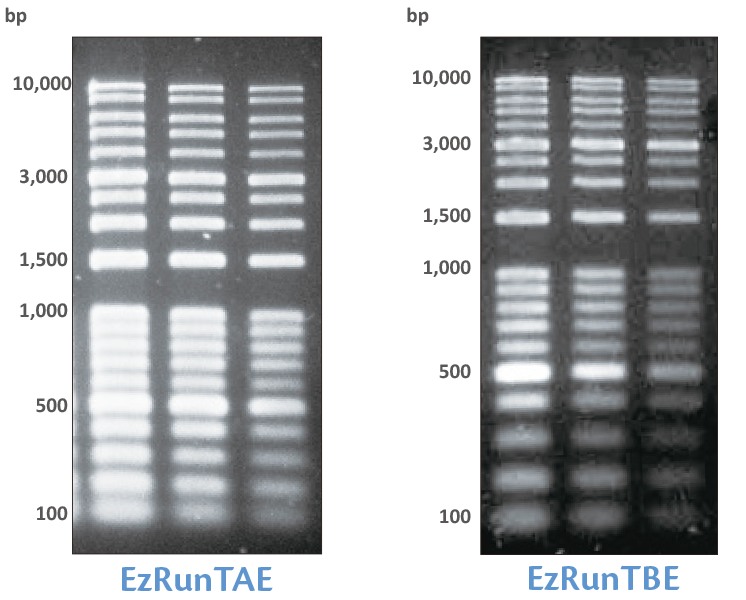
上図は、EzRunTAEまたはEzRunTBEバッファーを使用した1.5%アガロースゲルに分子量マーカーをアプライし、サブマージ ミニを使用して150Vで20分間電気泳動した結果を示しています。
染色にはEzFluoroStainDNAを用いて、Cyan LED励起で撮影しました。このように、TAEバッファーは高分子の分離、TBEバッファーは低分子の分離に適していることがわかります。
豆知識1:ローディングバッファーの色素
ローディングバッファーには、サンプルをウェルに均等に沈ませるためのグリセロール等の比重添加剤と、
サンプルに色を付けてゲルにアプライできているかを簡単に確認できるように色素が添加されています。
色素は電気泳動の進行具合を確認したり、泳動距離の目安にするためにも非常に重要ですが、添加されている色素によって電気泳動の速度が異なります。
どの色素を使用するかは、分離したいDNAの大きさや個人の好みによります(下表参照)。
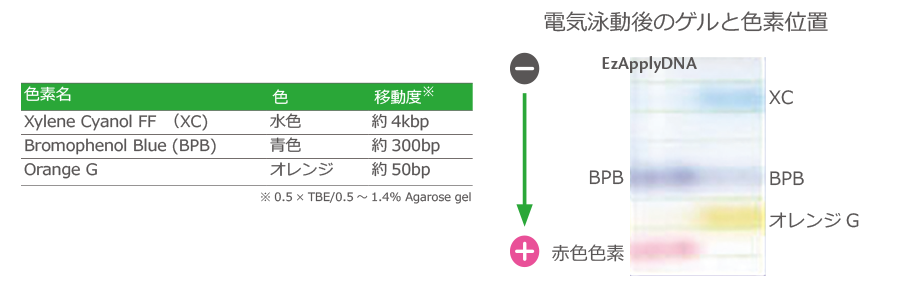
アトーのEzApplyDNAには、BPBと赤色色素の2種類が添加されています(上図参照)。
特に赤色色素はオレンジGよりも速く進むため、泳動先端の指標となり、うっかり流しすぎてしまった!なんて失敗がなくなります。
さらに比重添加剤としてFicollを使用しているため、グリセロール添加バッファーよりも、明瞭でシャープなバンドを得ることができます。
豆知識2:アガロースゲルの染色
エチジウムブロマイド(EtBr)などによる蛍光染色では、紫外線(UV)などの照射により蛍光物質が励起されて蛍光を発することで、バンドを可視化することができます。
蛍光物質は核酸に特異的に結合し、その結合量は核酸の分子量や濃度に依存しています。
つまり、分子量が大きく量が多いバンドはより強く光り、分子量が小さく量が少ないバンドは蛍光が弱くなります。
EtBrはゲルへの浸透が早く、短時間の染色でも十分な感度が得られます。
しかし発がん性物質である上に、励起にUVが必要であることから、EtBrによるゲル染色は非常に人体に危険な操作です。
さらにUV照射により核酸が分解されてしまうため、UVを照射し続けると蛍光は徐々に弱くなり、最終的にバンドは消失してしまいます。
アトーのDNA検出用の蛍光染色試薬EzFluoroStainDNAやEzPreStainDNA&RNAは、EtBrよりも低いバックグラウンドで高感度に検出が可能です。
励起光にCyan LEDや青色LEDが使用できるので、UVより安全に感度良く検出が可能です。
さらにLED光源での励起はUVのように核酸を分解しないので、ゲル切出し後の核酸の集率が向上するというメリットもあります。
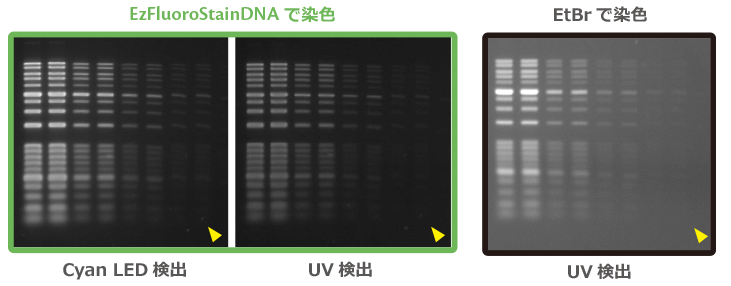
上図はEzFluoroStainDNAとEtBrで染色したゲルをCyan LEDまたはUVで検出した結果を示しています。
EzFluoroStainDNAで染色し、CyanLEDで励起したものが1番高感度にバンドが検出できていることがわかります。
資料ダウンロード

動画で紹介
サブマージ ミニの使用方法
関連製品
 バンドがシャープになる、DNA電気泳動用ローディング試薬です(BPB、赤色色素入り)。WSE-7050 EzRun TAE¥9,800
バンドがシャープになる、DNA電気泳動用ローディング試薬です(BPB、赤色色素入り)。WSE-7050 EzRun TAE¥9,800 トリス-酢酸系の電気泳動用バッファーです。WSE-7051 EzRunTBE¥5,800
トリス-酢酸系の電気泳動用バッファーです。WSE-7051 EzRunTBE¥5,800 トリス-ホウ酸系の電気泳動用バッファーです。WSE-1710 サブマージ ミニ¥43,800
トリス-ホウ酸系の電気泳動用バッファーです。WSE-1710 サブマージ ミニ¥43,800 電源搭載の小型サブマリン電気泳動装置です。DNAのアガロースゲル電気泳動等にご利用ください。WSE-1720 サブマージ マルチ¥63,800
電源搭載の小型サブマリン電気泳動装置です。DNAのアガロースゲル電気泳動等にご利用ください。WSE-1720 サブマージ マルチ¥63,800 電源搭載の小型サブマリン電気泳動装置です。DNAのアガロースゲル電気泳動等にご利用ください。WSE-7130 EzFluoroStain DNA¥19,800
電源搭載の小型サブマリン電気泳動装置です。DNAのアガロースゲル電気泳動等にご利用ください。WSE-7130 EzFluoroStain DNA¥19,800 電気泳動後のDNA検出用蛍光染色剤です。EtBrより高感度・安全にDNAを検出し、BlueLEDによる励起でより安全・高感度に検出できます。WSE-7135 EzPreStain DNA&RNA¥15,800
電気泳動後のDNA検出用蛍光染色剤です。EtBrより高感度・安全にDNAを検出し、BlueLEDによる励起でより安全・高感度に検出できます。WSE-7135 EzPreStain DNA&RNA¥15,800 ゲルや泳動バッファーに混ぜることで、DNAやRNAを蛍光染色しながら電気泳動可能な検出試薬です。
ゲルや泳動バッファーに混ぜることで、DNAやRNAを蛍光染色しながら電気泳動可能な検出試薬です。