






















テクニカルインフォメーション
Total Proteinによるノーマライズ
ウェスタンブロッティングはターゲットタンパク質の検出をするために、最もよく使用されている方法です。ターゲットタンパク質の発現量の変化や、サンプル間における発現量の比較を検出するためには、ターゲットタンパク質の量を何らかの方法で定量する必要があります。一般的にウェスタンブロッティングのデータはサンプル間やレーン間でのバラつきがあります。そこで、それらの影響を最小限にするために、ある指標(リファレンス)を利用して補正します(ノーマライズ)。これまでノーマライズといえば、ハウスキーピングタンパク質(HKP) の発現量がリファレンスとして利用されてきました。しかし最近になって、ハウスキーピングタンパク質の発現量が組織や細胞種によって、また細胞周期や発生段階によって必ずしも一定ではないこと、その発現量もターゲットタンパク質のものとはかけ離れていることが指摘されるようになり、ノーマライズに使用する場合は十分注意する必要があるといわれるようになりました。そんな中、ハウスキーピングタンパク質に変わってノーマライズに使用できるリファレンスとして、Total protein( TP、トータルタンパク質、総タンパク質) が注目されてきています。今回は、アトー製品などを使用したトータルタンパク質 によるデータノーマライズの方法をご紹介します。
ノーマライズとは
タンパク質のサンプルは不均一な上に、同じサンプルを同じ量だけアプライして電気泳動をしたとしても、レーン間の差は生じます。どんなに注意をしても、転写ムラや抗体反応のバラつきなども避けられません。そこで精度良く、再現性良く、定量的にウェスタンブロッティングを行う上で、ノーマライズというプロセスは必要不可欠になります。ウェスタンブロッティングのノーマライズでは、ターゲットタンパク質の輝度値を、リファレンスの輝度値から算出された各レーンの相対値(リファレンス比)で割って補正します。
ターゲットタンパク質の検出
① 電気泳動
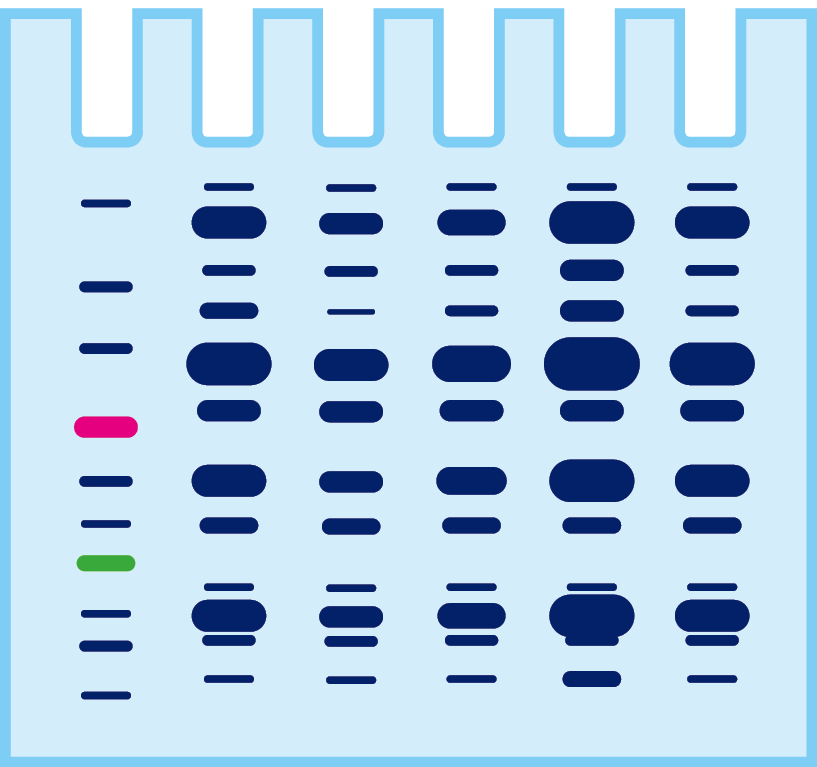
サンプル中に含まれるタンパク質は?
電気泳動をするとサンプル中に含まれるタンパク質がバンドに分かれます。しかし、ターゲットタンパク質が、どのバンドなのかは判別できません。
② ウェスタンブロッティング
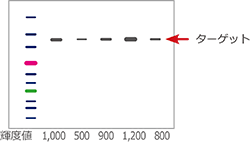
ターゲットタンパク質は発現してるの?
ウェスタンブロッティングをするとターゲットタンパク質のバンドと量(輝度値)が分かります。分子量マーカーの移動度から、ターゲットタンパク質の分子量も推定できます。しかし、どのサンプルで最も多く発現しているのかは判断できません。
③ データの解析
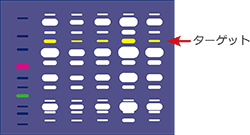
ターゲットタンパク質のバンドはどれ?
電気泳動パターンとウェスタンブロッティングの検出バンドを重ねると、どのバンドがターゲットタンパク質なのかが判別できます。各レーンのトータルタンパク質の情報をリファレンスとして、サンプル中にどのくらい含まれるのか、どのサンプルで最も多く発現しているのかなども解析可能になります。
なぜノーマライズは必要?
下記の影響を最小限にするためノーマライズは必要です
① サンプル自体の不均一さ
生物種や組織・発生ステージによる違い、抽出方法などにより影響を受けます。
② アプライ量の不均一さ
タンパク質定量の方法や抽出溶媒の影響、アプライするときの手技などにより影響を受けます。
③ 転写効率・検出効率・抗体反応の不均一さ
転写バッファーや転写方法、手技、抗体のタイターや希釈率や反応時間などにより影響を受けます。
④ 複数のブロット間での比較
多検体を比較するときなど複数のブロットの発現量を比較するときに基準が必要になります。
ノーマライズのリファレンスには?
データノーマライズはターゲットタンパク質の発現量をリファレンスにより補正します。
| リファレンスの種類 | |
|---|---|
|
① トータルタンパク質
|
② ハウスキーピングタンパク質
|
※ただし、ハウスキーピングタンパク質をリファレンスに使用するときは下記の注意が必要といわれています。
- 実験条件によりリファレンスの発現量が影響されないこと
- リファレンスの発現量とターゲットタンパク質の発現量はかけ離れていないこと
- リファレンスの検出がターゲットタンパク質の検出を妨げないこと
ノーマライズによる定量
リファレンスの輝度値のレーン間での比(『リファレンス比』)を算出し、これをばらつきの補正係数とします。
[リファレンス比(ref.比)]= [各リファレンスの輝度値] ÷ [最初の番号のレーンのリファレンスの輝度値]
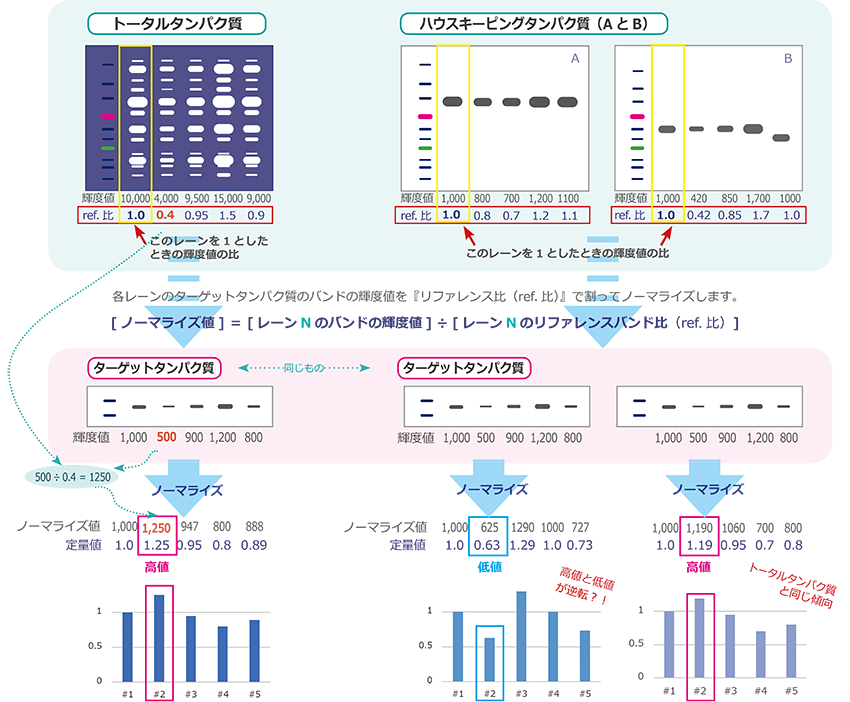
① トータルタンパク質によるノーマライズ
PVDF膜に転写されたタンパク質量(トータルタンパク質)をリファレンスとしてノーマライズした例です。トータルタンパク質は、ある特定のタンパク質の発現量ではないため、普遍的な指標といえます。抗体反応や転写ムラによる影響や、組織の違いなどによる影響も最小限に抑えられます。上記は最もターゲットタンパク質のシグナルが弱かったサンプルが、ノーマライズした結果、最も多く発現している例を示しています。
② ハウスキーピングタンパク質によるノーマライズ
GAPDHやアクチンなどのハウスキーピングタンパク質をリファレンスとしてノーマライズした例です。ハウスキーピングタンパク質は比較的安定に発現していますが、組織により発現量が異なる場合や、発現量がターゲットタンパク質よりも著しく高い場合、抗体の反応性が良く過剰に検出される場合は、正確に補正できないことがあります。上記はハウスキーピングタンパク質が過剰に検出される例(A)と理想的に検出される例(B)のイメージです。極端な例ですが、発現量が高いサンプルと低いサンプルが逆転する可能性を示しています。
実験の流れ
下図はトータルタンパク質でノーマライズするための実験フローを示しています。トータルタンパク質 でノーマライズする場合、転写されたPVDF膜上のタンパク質バンドを視覚化する必要があります。PVDF膜上のタンパク質を検出する方法は大きく分けて①サンプル標識、②ステインフリーゲル、③転写後の膜染色があります。
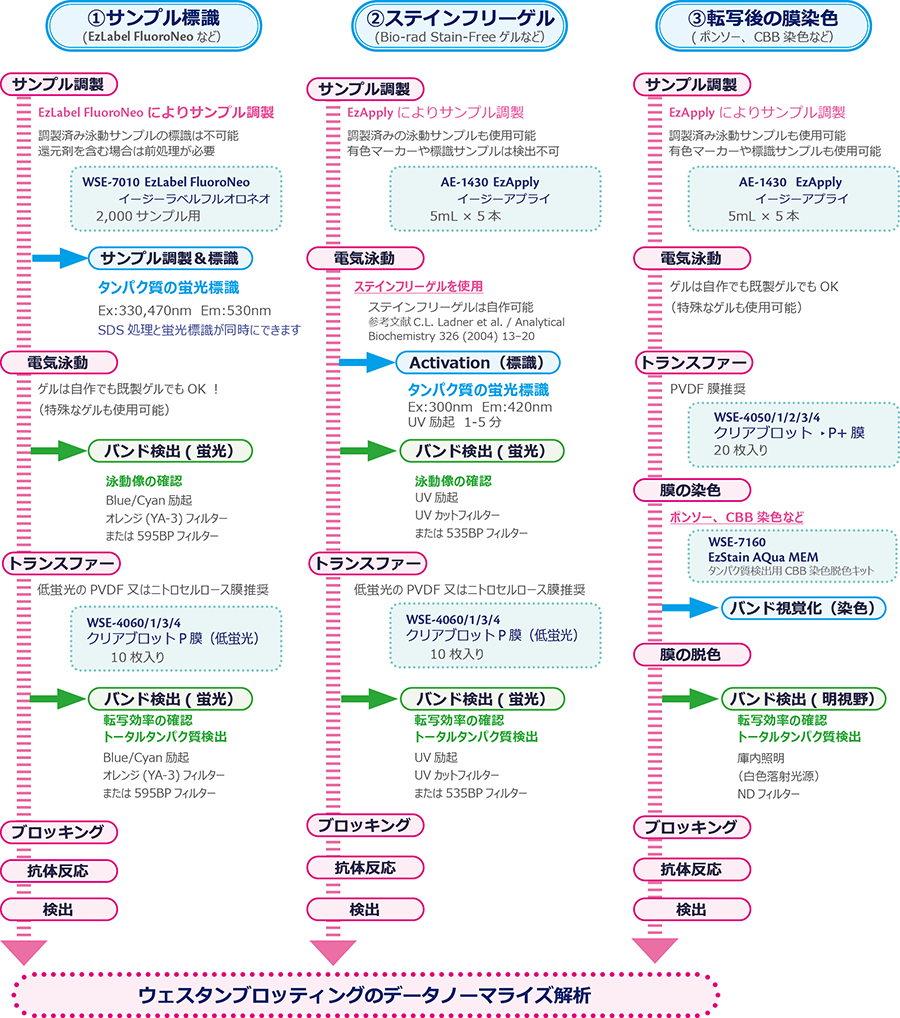
EzLabel FluoroNeoを使用したトータルタンパク質の検出
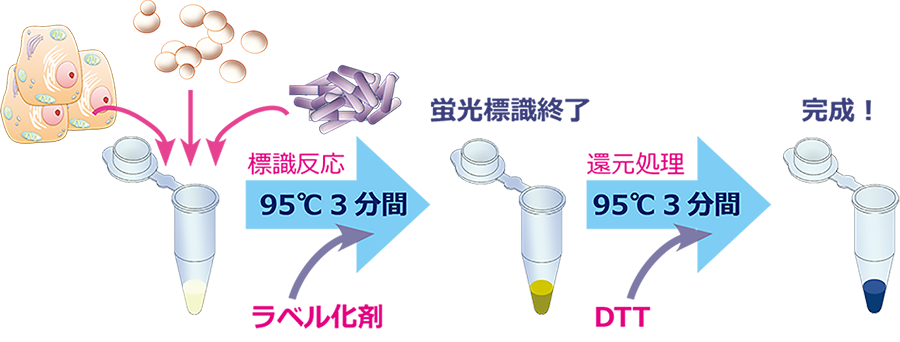
1.サンプル調製(EzLabel FluoroNeo)
EzLabel FluoroNeoはSDSサンプルバッファーの代わりに使うだけで、タンパク質の蛍光標識と電気泳動用サンプル調製ができます。
電気泳動後のゲルは、染脱色不要で、すぐにバンドが蛍光検出できます。
実験材料
- EzLabel FluoroNeo(WSE-7010)
- 組織・細胞・細菌など
- マイクロ遠心チューブ、チップなど
- 遠心機、マイクロピペットなど
実験方法
1.EzLabel FluoroNeoを使用して電気泳動サンプルを調製します。40 µLのタンパク質サンプルにキット添付の10 μLのSample buffer (5x conc.) とLabeling reagent を0.5 μL添加して混合します。
2.95℃で3分間加熱します(煮沸でもOK)。
3.2.の混合液にReducing agent (DTT)を2μL 添加して混合します。
4.95℃で3分間加熱します(煮沸でもOK)。
※作製したサンプルは-20℃で遮光保存可能です。
※サンプル中に還元剤が含まれると蛍光標識反応を著しく妨げる原因となります。そのため標識後に還元処理(ステップ3~4)をします。
2.電気泳動
EzLabel FluoroNeoで標識した電気泳動サンプルは、一般的なゲル、泳動バッファー、泳動条件で分離できます。
ここでは既製ゲル(ePAGEL-HR)を使用した、高速電気泳動に関してご紹介します。

実験材料
- WSE-7010EzLabel FluoroNeoで調製した泳動サンプル
- 既製ゲル(ePAGEL-HRなど)
- 泳動バッファー(AE-1410 EzRunなど)
- 電気泳動装置、電源、チップなど
実験方法
1.ePAGEL-HRをパジェランAce(電源付電気泳動装置)にセットします。
2.1レーンあたり5~10μLのサンプルをアプライします。
※サンプル濃度は精製タンパク質では100ng~1μg/lane、抽出液では1~50μg/laneが適当です。
3.スタートボタンを押して電気泳動を開始します。
※High mode (24W)で約35分間、もしくはStandard mode (20mA/gel)で約80分間泳動します。
※ 外部電源を使用する場合は、300Vで約35分間、もしくは150Vで 約75分間泳動します。
4.泳動先端(色素ライン)がゲルの下端から5-10 mm上まで泳動されたら、出力を止めて終了します。
※サンプルの溶媒にTrisなどのアミノ基が含まれると、非特異的なシグナルが出る原因になります
(ゲルの蒸留水洗浄により消えます)。
3.電気泳動パターンの確認(蛍光検出)
EzLabel FluoroNeoを使用した電気泳動後のゲルは、すぐにBlue LEDあるいはCyanで励起することにより、バンドパターンを確認できます。

実験材料
- 泳動後のゲル(WSE-7010EzLabel FluoroNeo使用)
- LuminoGraph III などの撮影装置
- Cyan (CyanoView) などの光源とフィルター
実験方法
1.泳動終了後のゲルを泳動槽から外し、ゲルプレートを外さずに、水道水でガラス表面を軽く洗浄し、水気をペーパータオルで拭きとります。
2.撮影装置と光源、フィルターを準備します。
3.ゲルをプレートごと光源照射面に置いて撮影します。[Ex: 330 (UV), 470 nm, Em: 530 nm]
| 撮影装置 | 光源 | フィルター |
|---|---|---|
| LuminoGraph I/IIなど | VariRays (Blue)もしくはCyanoView | YA-3(560LPF)もしくはオレンジフィルター |
| LuminoGraph III | Blue LEDもしくはCyan光源 |
BPF595もしくはBPF535 |
4.転写
通常の転写条件で転写できます。PVDF膜は、自家蛍光の低い蛍光検出用のPVDF膜の使用が望ましいですが、一般的なPVDF膜でも検出可能です。特にBlue LED光源で検出する場合は、自家蛍光の影響が低減できます。ここでは高速転写バッファー(EzFastBlot)を使用した、高速転写に関してご紹介します。

実験材料
- 泳動後のゲル(WSE-7010 EzLabel FluoroNeo使用)
- 転写バッファー(AE-1465 EzFastBlotなど)
- PVDF膜(クリアブロットP膜(低蛍光)など)
- ブロッティング装置(PoweredBLOT 2Mなど)
- 遠心機、マイクロピペットなど
実験方法
1.あらかじめPVDF膜をメタノールで親水化処理し(5秒)、EzFastBlotに浸漬します(15分以上振とうします)。
2.蛍光検出後のゲルをEzFastBlotで洗浄し、陽極側(下)からろ紙(0.9mm厚)2~3枚、PVDF膜、ゲル、ろ紙2~3枚の順に重ねます。ろ紙は重ねる直前にEzFastBlotに充分浸漬します。
※空気を専用ローラーを使って押し出し、ろ紙、膜、ゲルを密着させます。
3.スタートボタンを押してトランスファーを開始します。
※Rapid modeで10~15分通電します。
※ 外部電源を使用する場合は、24Vで約10~15分間、もしくは12Vで 約30分間転写します。
| 転写バッファーと転写条件 | EzBlot | EzFastBlot | EzFastBlotHMW | EzRun TG(Towbin法) |
|---|---|---|---|---|
| 型式 | AE-1460 | AE-1465 | WSE-7210 | WSE-7055 |
| 転写条件 | 2 mA/cm2 60 min (~40V) |
20~25V c.v. 5~15 min (500mA) |
25V c.v. 15~30 min (500mA/gel) |
2 mA/cm2 60 min (~40V) |
5.転写効率の確認(蛍光検出)
EzLabel FluoroNeoで調製したサンプルは、転写後のPVDF膜上のバンドもゲルと同様に蛍光検出できます。
EzLabel FluoroNeoの蛍光強度は、タンパク質濃度と相関しているため、PVDF膜上のタンパク質バンドのシグナル強度をトータルタンパク質としてノーマライズに利用することが可能です。
ノーマライズに関しては【ノーマライズの方法】を参照してください。

実験材料
- 転写後のPVDF膜(EzLabel FluoroNeo使用)
- LuminoGraph III などの撮影装置
- Cyan (CyanoView) などの光源とフィルター
実験方法
1.転写終了後のPVDF膜を蒸留水などで軽く洗浄します。
2.撮影装置と光源、フィルターを準備します。
※PVDF膜は汚れないように十分注意して取り扱ってください。ピタットクリアに挟むと、蛍光検出に影響を与えずに、汚れから保護できます。
3.PVDF膜を光源照射面に置いて撮影します。
※PVDF膜は乾燥した方が、蛍光シグナルを強く明瞭に検出できます。乾燥したPVDF膜はメタノールで親水化処理をした後に、ブロッキング、抗体反応に進んでください。
| 撮影装置 | 光源 | フィルター |
|---|---|---|
| LuminoGraph I/IIなど | VariRays (Blue)もしくはCyanoView | YA-3(560LPF)もしくはオレンジフィルター |
| LuminoGraph III | Blue LEDもしくはCyan光源 |
BPF595もしくはBPF535 |
ステインフリーゲルを使用したトータルタンパク質の検出
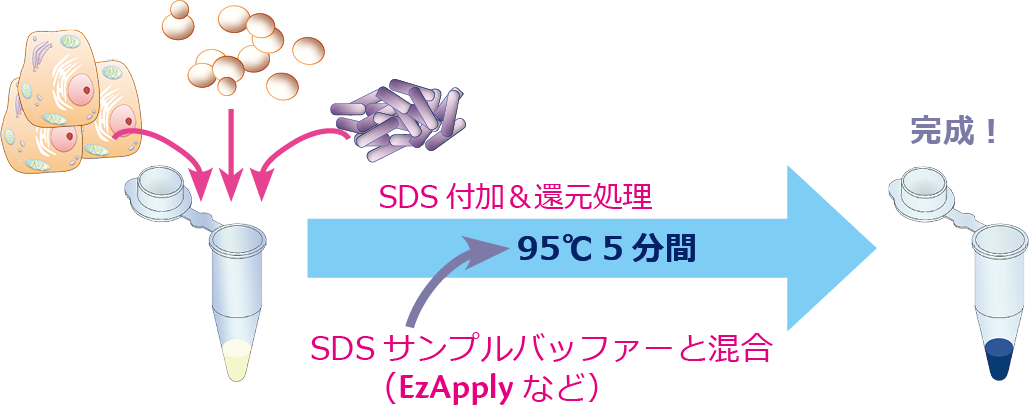
1.サンプル調製(EzApply)
ステインフリーゲルを使用する場合も、電気泳動サンプルは通常の方法で調製します。有色マーカーは検出できないので、無色マーカーを併用します。
実験材料
- 組織・細胞・細菌など
- SDSサンプルバッファー(AE-1430 EzApply)
- マイクロ遠心チューブ、チップなど
- 遠心機、マイクロピペットなど
実験方法
1.50μLのサンプルに50μLのEzApply (2×濃度、DTT添加済み)を加えて混合します。
2.小型ブロックインキュベータWSC-2610 MyMiniBlockで95℃5分間加熱します(煮沸でもOK)。
3.15,000rpmで5分間遠心し(しなくてもOK)上清を回収します。
※作製したサンプルは-20℃で保存可能です。
2.電気泳動
ステインフリーゲルを使用して泳動します。プレキャストゲルを使用する場合は、取扱説明書に従ってください。ここでは、自作ステインフリーゲルを作製し、泳動する方法を紹介します。
ステインフリーゲル作製方法の詳細は下記Ladnerらの文献を参照してください。
【参考文献】
C.L. Ladner et al., Analytical Biochemistry 326 (2004) 13–20
Visible fluorescent detection of proteins in polyacrylamide gels without staining
実験方法
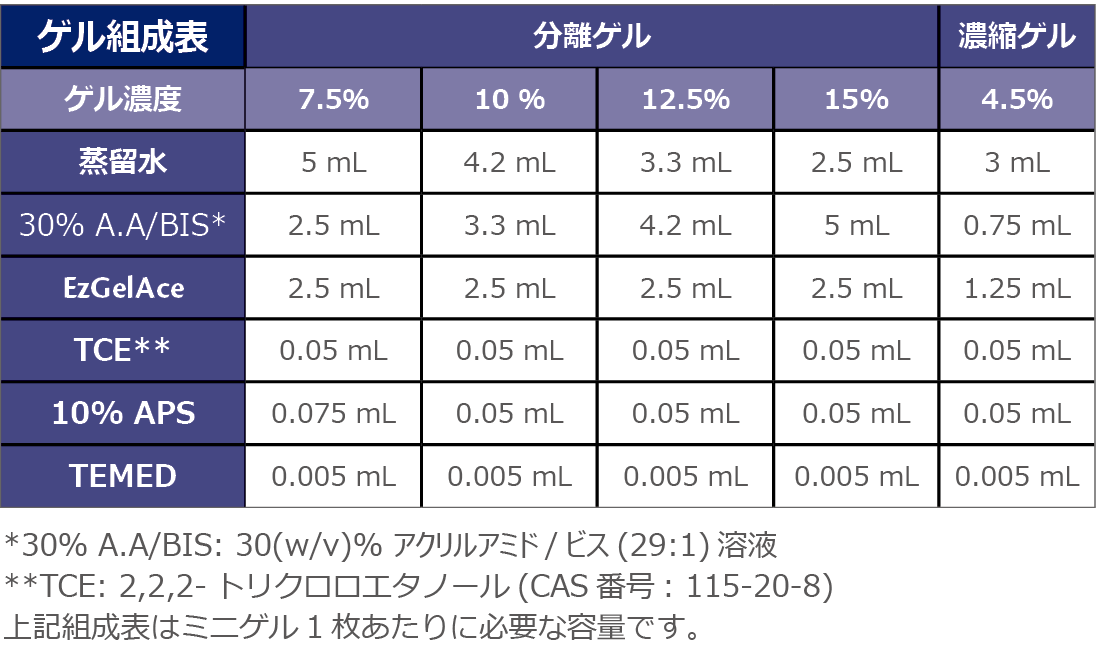
1.ゲル作製器を組み立てます。組み立て方法はご使用になる作製器の取扱説明書に従ってください。
2.上記のゲル組成表を参照して、分離ゲルと濃縮ゲルのゲル溶液をAPSとTEMED以外の試薬を混合して調製します。重合開始直前にAPSとTEMEDを添加して、ゲルを作製します。
3.ゲルをパジェラン Ace(電源付電気泳動装置)にセットします。
4.1レーンあたり5~10μLのサンプルをアプライします。
※サンプル濃度は精製タンパク質では100ng~1μg/lane、抽出液では1~50μg/laneが適当です。
5.スタートボタンを押して電気泳動を開始します。
※High mode (24W)で約35分間、もしくはStandard mode (20mA/gel)で約80分間泳動します。
※ 外部電源を使用する場合は、300Vで約35分間、もしくは150Vで 約75分間泳動します。
6.泳動先端(色素ライン)がゲルの下端から5~10 mm上まで泳動されたら、出力を止めて終了します。
3.検出
電気泳動後のゲルは、すぐには蛍光検出できません。タンパク質を可視化するために、ゲルを直接UV照射装置の上に置き、約1分間照射します。UV波長は312nmを使用します。
いったんUV照射で可視化したゲルの蛍光は比較的安定なので、蒸留水等でリンスしてゲルの状態を整えてから撮影すると、きれいな画像が得られ、電気泳動パターンが確認できます。

実験材料
- 泳動後のゲル(ステインフリーゲル使用)
- LuminoGraph IIIなどの撮影装置
[UV光源(312nm) とフィルター]
実験方法
ゲルの可視化
1.泳動終了後のゲルを、ゲルプレートを外さずに水でガラス表面を軽く洗浄し、ペーパータオルで水気を拭きとります。
2.UV照射装置の上に蒸留水を1~5mL滴下します。
3.ゲルをプレートから外し、UV照射装置に滴下した水滴の上に載せます。
※ゲルをリンス・洗浄しないでください。シグナルが弱くなります。
※UVの波長が異なると可視化されない原因になります。
※UV照射光源を直視・接触しないようにご注意ください。
※ゲルを可視化するための工程なので、照射面にゲルを整えて置く必要はありません。またゲルが破れやすいので注意していください。
4.UV照射装置を点灯し、ゲルを約1分間、照射します。
5.UV照射後、蒸留水等でリンスしてゲルの状態を整える。
ゲルの撮影
6.撮影装置のUV光源とフィルターをセットします。
7.ゲルをUV照射面もしくは専用のトレイに置いて撮影します。
※UV照射面をきれいにしてから撮影してください。
※UV照射面の水垢汚れはくえん酸水溶液を使用すると落とせます。
| 撮影装置 | 光源 | フィルター |
|---|---|---|
| LuminoGraph I/IIなど | UV光源(312nm) | SWPパスフィルター(UV/近赤外カット) |
| LuminoGraph III | UV光源(312nm) |
BPF535 |
4.転写
通常の転写条件で転写できます。PVDF膜は、自家蛍光の低い蛍光検出用のPVDF膜の使用が望ましいです。
ここでは高速転写バッファー(EzFastBlot)を使用した、高速転写に関してご紹介します。
実験材料
- 泳動後のゲル(ステインフリーゲル使用)
- 転写バッファー(AE-1465 EzFastBlot など)
- PVDF膜(クリアブロットP膜(低蛍光)など)
- ブロッティング装置(PoweredBLOT 2Mなど)
- 遠心機、マイクロピペットなど
実験方法
1.あらかじめPVDF膜をメタノールで親水化処理し(5秒)、EzFastBlotに浸漬します(15分以上振とうします)。
2.蛍光検出後のゲルをEzFastBlotで洗浄し、陽極側(下)からろ紙(0.9mm厚)2~3枚、PVDF膜、ゲル、ろ紙2~3枚の順に重ねます。ろ紙は重ねる直前にEzFastBlotに充分浸漬します。
※空気を専用ローラーを使って押し出し、ろ紙、膜、ゲルを密着させます。
3.スタートボタンを押してトランスファーを開始します。
※Rapid modeで10~15分通電します。
※ 外部電源を使用する場合は、24Vで約10~15分間、もしくは12Vで 約30分間転写します。
| 転写バッファーと転写条件 | EzBlot | EzFastBlot | EzFastBlotHMW | EzRun TG(Towbin法) |
|---|---|---|---|---|
| 型式 | AE-1460 | AE-1465 | WSE-7210 | WSE-7055 |
| 転写条件 | 2 mA/cm2 60 min (~40V) |
20~25V c.v. 5~15 min (500mA/gel) |
25V c.v. 15~30 min (500mA/gel) |
2 mA/cm2 60 min (~40V) |
5.転写効率の確認(蛍光検出)
可視化処理をしたステインフリーゲルは、転写後のPVDF膜上のバンドもゲルと同様に蛍光検出できます。
PVDF膜上のタンパク質バンドのシグナル強度をトータルタンパク質としてノーマライズに利用することが可能です。
ノーマライズに関しては【ノーマライズの方法】を参照してください。
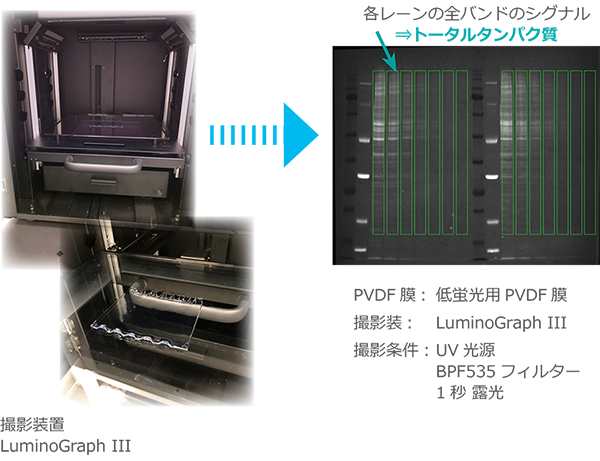
実験材料
- 転写後のPVDF膜(ステインフリーゲル使用)
- LuminoGraph IIIなどの撮影装置
[UV光源(312nm) とフィルター]
実験方法
1.転写終了後のPVDF膜を蒸留水などで軽く洗浄します。
2.撮影装置のUV光源、フィルターを準備します。
3.PVDF膜をUV照射面もしくは専用のトレイに置いて撮影します。
※PVDF膜は汚れないように十分注意して取り扱ってください。
※UV照射により励起する場合、PVDF膜は濡れている方が、バックグラウンドが低くなり、バンドを明瞭に検出できます。撮影中に乾燥しないようにご注意ください。
※ピタットクリアやラップに挟むと、バックグラウンドが高くなる場合がありますので、ご注意ください。
| 撮影装置 | 光源 | フィルター |
|---|---|---|
| LuminoGraph I/IIなど | UV光源(312nm) | SWPパスフィルター(UV/近赤外カット) |
| LuminoGraph III | UV光源(312nm) |
BPF535 |
トータルタンパク質によるノーマライズの方法
1.トータルタンパク質によるノーマライズ
EzLabelFluoroNeoやステインフリーゲルなどを使用して検出したトータルタンパク質をリファレンスとしてノーマライズします。
リファレンスにはPVDF膜上の各レーンの転写されたタンパク質の総量(輝度値)を使用します。
2.ノーマライズの手順
EzLabelFluoroNeoを使用する場合も、ステインフリーゲルを使用する場合も、同様の方法で画像を解析します。
ノーマライズに必要な画像はPVDF膜上に転写されたトータルタンパク質のバンド(蛍光像または色素染色した場合は明視野像)と抗体反応後のターゲットタンパク質のバンド(発光像あるいは蛍光像など)です。
まずPVDF膜上のトータルタンパク質の各レーンのバンド全体のシグナル強度を、続いてターゲットバンドのシグナル強度を定量します。
ここでは画像解析ソフトCS Analyzer 4を使用して解析する方法を紹介します。
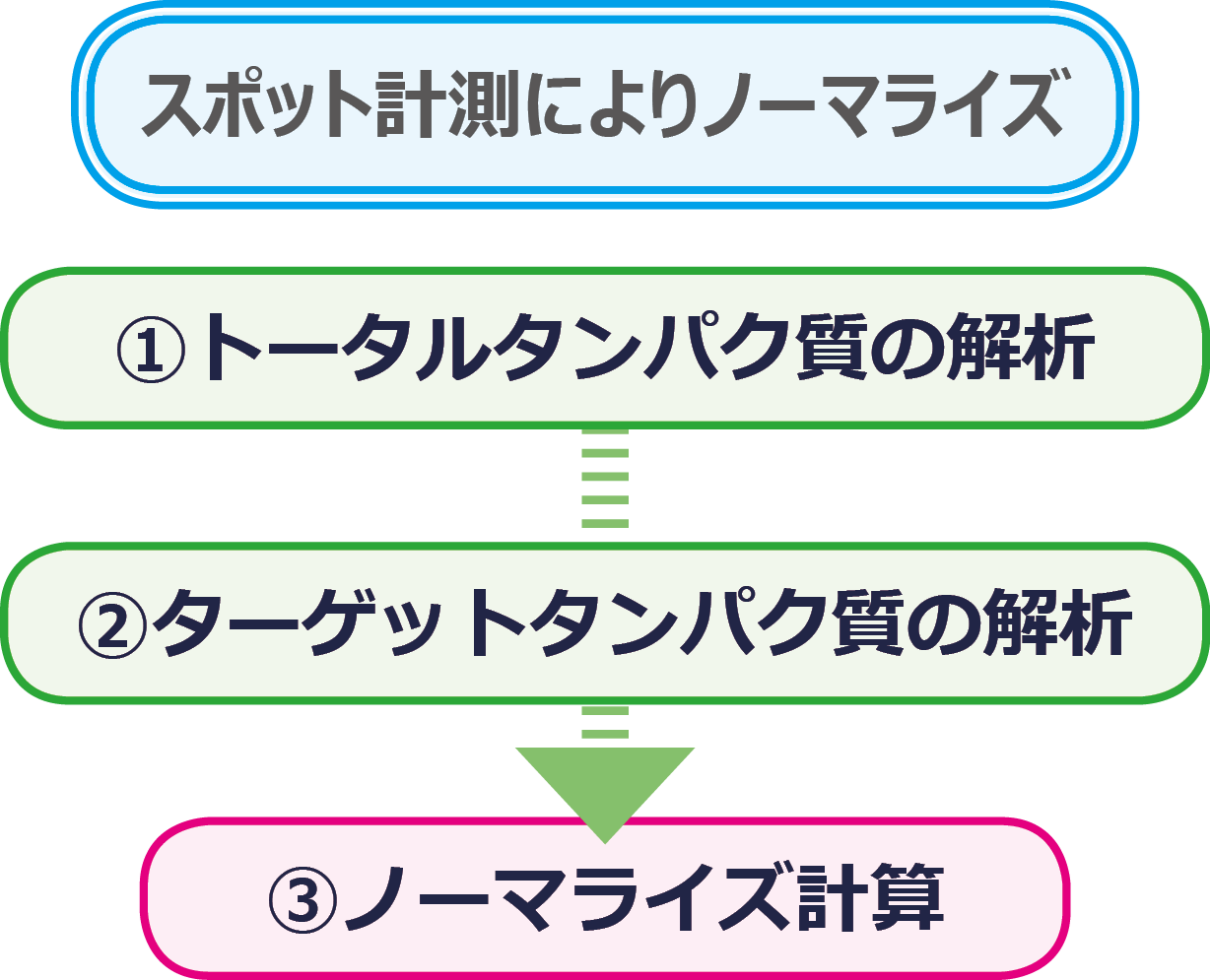
解析手順としては ①PVDF膜上のトータルタンパク質検出画像のスポット解析→②ターゲットタンパク質の検出画像のスポット解析→③ノーマライズ計算の手順で行います。
3.トータルタンパク質の解析
まずノーマライズ換算するためのリファレンスとなるトータルタンパク質を検出した蛍光撮影画像をスポット解析します。
① 各レーンの輪郭を囲む

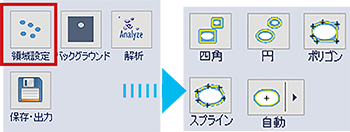 |
各レーンの輪郭を『領域設定』⇒『四角』あるいは『ポリゴン』などを選択して囲みます。
|
② バックグラウンドの設定
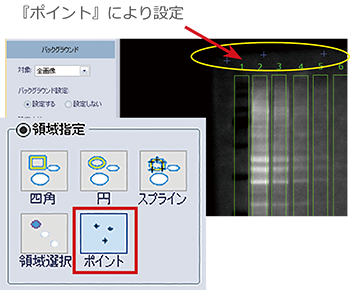
①で囲んだ領域以外のPVDF膜の余白部分にバックグラウンドを設定します。
『四角』や『領域選択』でスポットと同様の領域を設定する、または『ポイント』によりレーン以外の余白に複数のポイントを設定する、などの方法があります。
③ 解析およびデータ保存

解析ボタンをクリックしてバンドのシグナル強度を数値化し、さらに『解析情報保存』ボタンをクリックして解析結果を保存します。
解析後のデータはCSV形式でも出力できます。
4.ターゲットタンパク質の解析
次に抗体反応によりターゲットタンパク質を検出した発光(蛍光など)撮影画像をスポット解析します。解析方法はトータルタンパク質の解析方法と同様です。
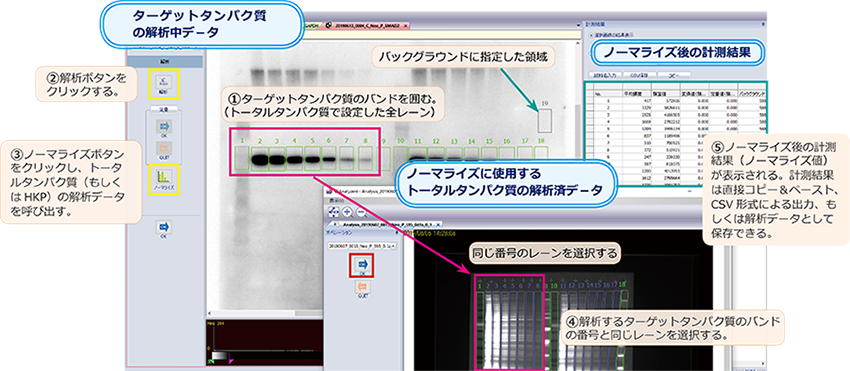
① バンドの輪郭を囲む
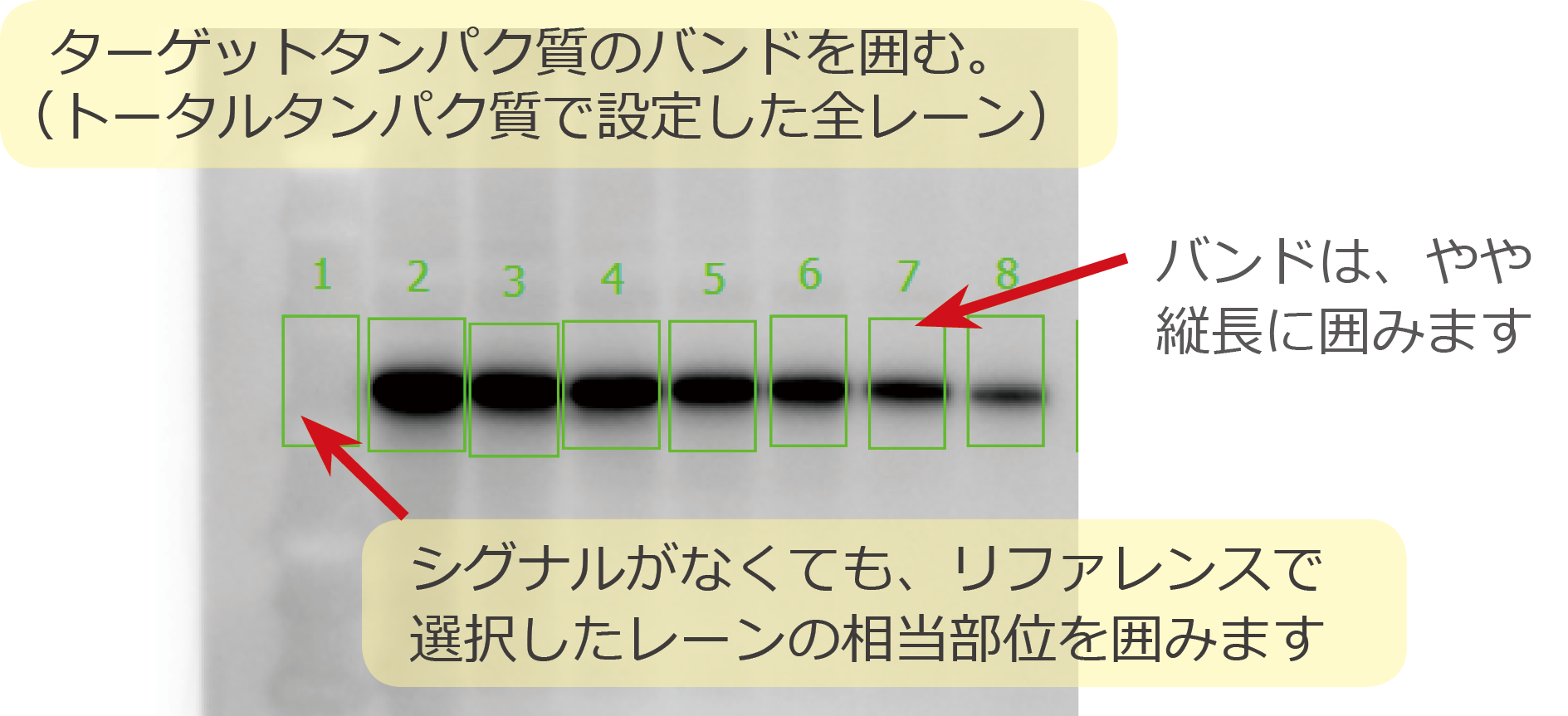
スポット解析により、ターゲットタンパク質のバンドの輪郭を『四角』あるいは『ポリゴン』を選択して囲みます。
特に発光撮影像はコントラストによりバンドの強さと大きさが影響されますので、ある程度コントラストを上げた状態でバンドを囲むようにします。
また、このとき、トータルタンパク質解析で設定したレーンの番号と解析する対象のバンドの番号は同じにしてください。
ターゲットのバンド番号とノーマライズするレーン番号がずれると、バンド番号と同じ番号の別レーンのデータで誤った補正がされることになります。
② バックグラウンドの設定
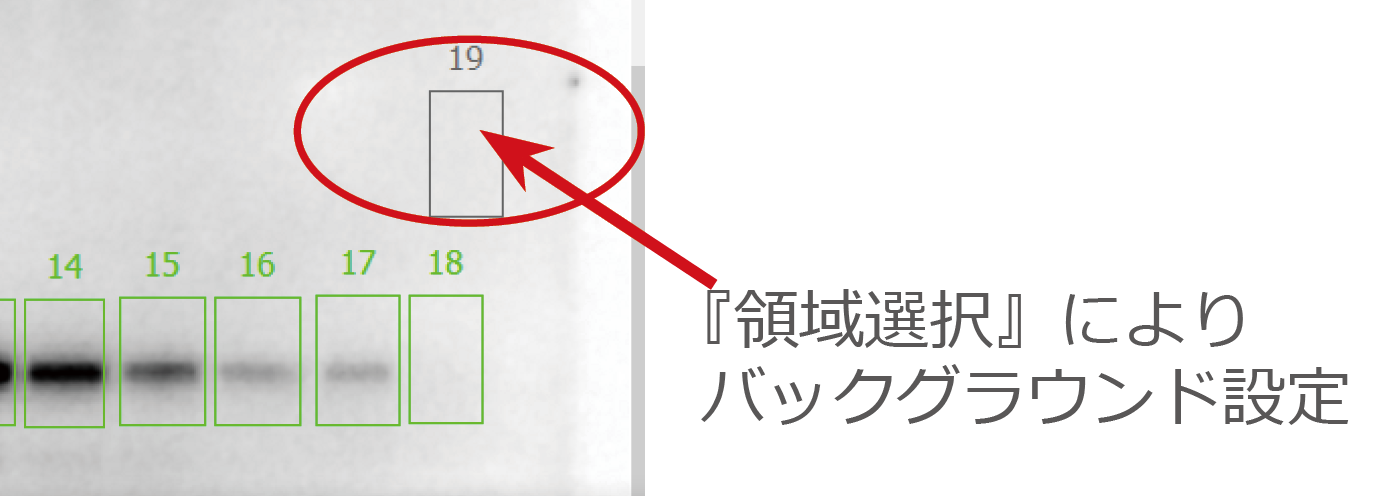
①で囲んだ領域以外のPVDF膜の余白部分にバックグラウンドを設定します。
『四角』や『ポリゴン』でスポットと同様の領域を囲むか、『ポイント』を利用してレーン以外の余白に複数のポイントを設定します。
③ ノーマライズ解析
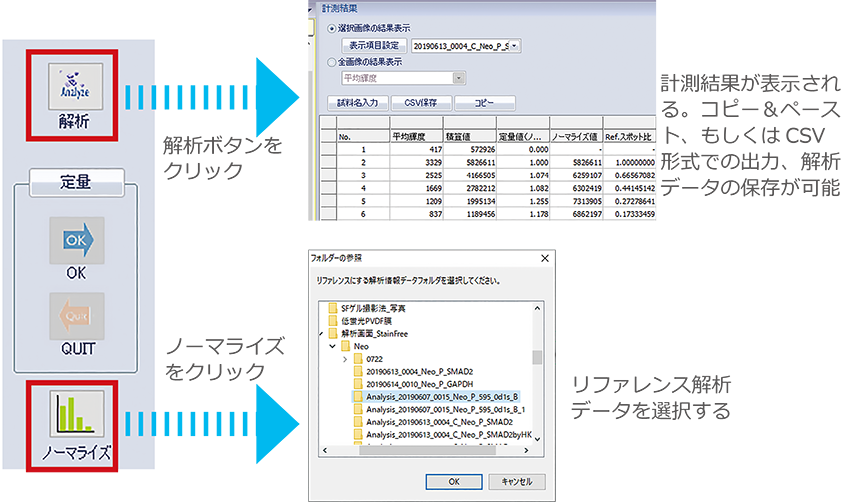 |
ターゲットタンパク質のバンドを囲み、バックグラウンドを設定したら、『解析』ボタンをクリックしてシグナル強度を数値化します。
『ノーマライズ』ボタンをクリックして、リファレンスタンパク質(トータルタンパク質)の解析データを呼び出します。 |
リファレンスタンパク質の解析データが表示されますので、ノーマライズするバンドと同じ番号のトータルタンパク質のレーンをクリックして選択し『OK』します。
ノーマライズの計測結果は、コピ-&ペースト、CSV形式での出力、もしくは解析データとして保存できます。
④ ノーマライズ値の定量解析
ノーマライズ結果を相対値として換算する場合は『定量』ボタンをクリックします。基準となるバンドをクリックして選択し『OK』します。
『ノーマライズ値』を選択し、『1(もしくは任意の数)』と入力します。
ノーマライズ値が相対値として換算されますので、コピ-&ペースト、CSV形式による出力、もしくは解析データとして保存します。
ノーマライズ解析結果
1.トータルタンパク質とハウスキーピングタンパク質によるノーマライズ定量結果の比較
下図はEzLabelFluoroNeoでラベルしたHeLa細胞抽出液からウェスタンブロッティングによりSMAD2タンパク質を検出したデータです。
1/2希釈系列したサンプル中のSMAD2タンパク質の発現量を、トータルタンパク質およびハウスキーピングタンパク質でノーマライズした結果を示しています。
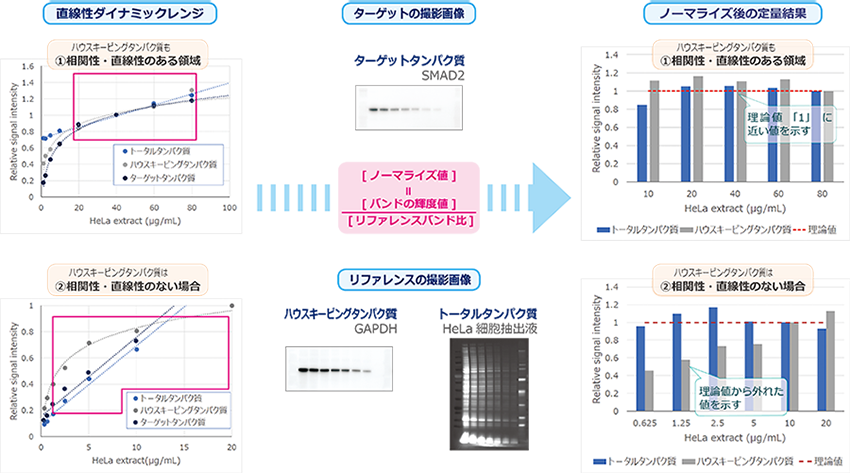
上図①のようにターゲットタンパク質とリファレンスタンパク質の検量線が直線で相関性が高い場合は、トータルタンパク質でもハウスキーピングタンパク質でも、
ノーマライズした結果は理論値の1に近い値を示します。
しかし一般的に②のようにハウスキーピングタンパク質は発現量が豊富なため、ターゲットタンパク質とは検量線の直線性ダイナミックレンジが異なります。
そのためハウスキーピングタンパク質によりノーマライズした結果は理論値から外れる場合があります。
2.EzLabel FluoroNeoとステインフリーゲルによるサンプル標識の比較
EzLabelFluoroNeoとステインフリーゲルにより検出したバンドを、ゲル、転写後のPVDF膜の蛍光検出像(膜_蛍光)とCBB染色像(膜_CBB) で比較した結果を示しています。
バンドを比較しやすいように、各イメージをグレースケールで表示しています。
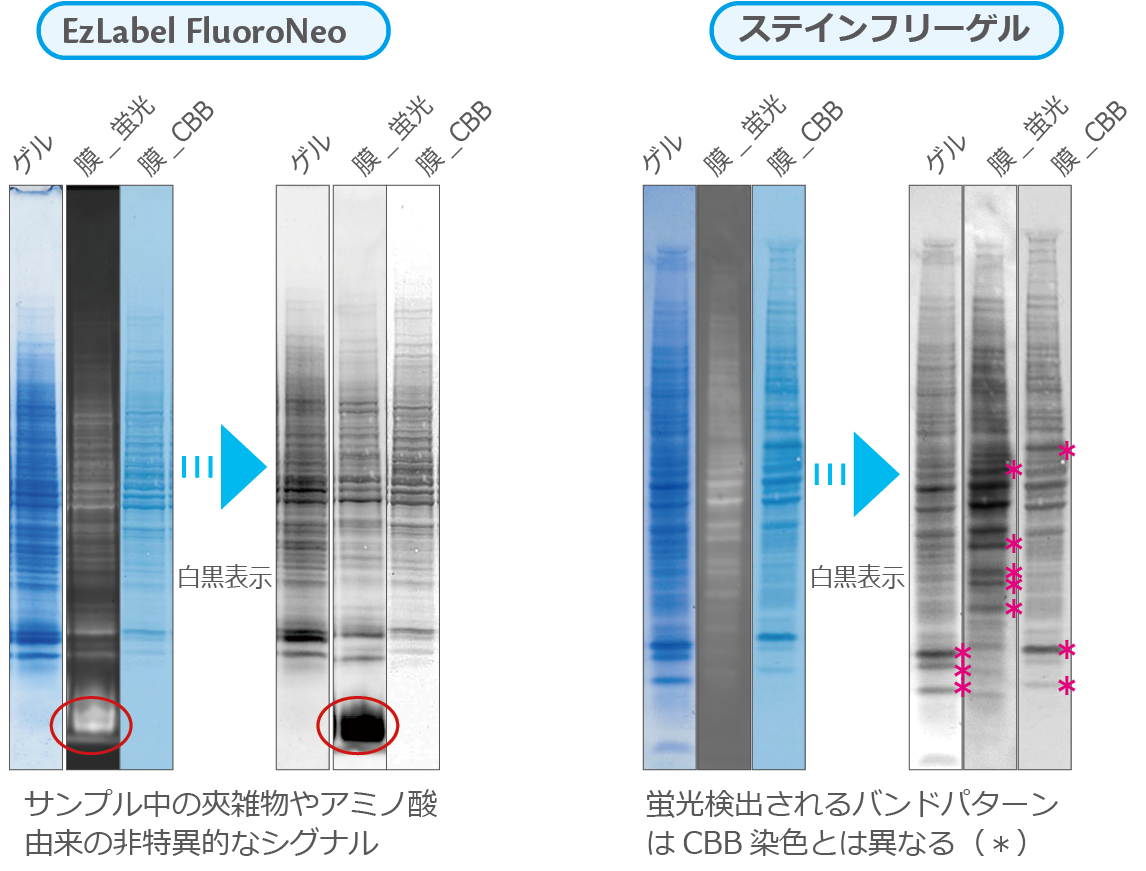
『膜_蛍光』のレーンは、EzLabelFluoroNeoもしくはステインフリーゲルの蛍光色素で標識されたバンドを検出した結果を示しています。
EzLabelFluoroNeoで検出すると(上図)、泳動先端にサンプル中のアミノ酸などに由来する非特異的なシグナルが見えますが(赤丸で表示)、
検出されるバンドの数やシグナルの強さは、ほぼ同等です。EzLabelFluoroNeoは第1、第2アミンが標識されるため、ほぼ全てのタンパク質が同等に標識されるためです。
一方、ステインフリーゲルはアミノ酸のうちトリプトファンが標識されるため、トリプトファンの有無や数によりシグナルの強さが変わります(上図)。
そのため、蛍光検出とCBB検出でバンドパターンがやや異なります。
*で示したバンドは、蛍光検出とCBB検出で検出感度が顕著に異なるバンドを示しています。
資料ダウンロード
関連製品
 電気泳動やウエスタンブロッティングの試料調製をしながらタンパク質やポリペプチドを蛍光標識する試薬キットです。AE-1430 EzApply¥9,800
電気泳動やウエスタンブロッティングの試料調製をしながらタンパク質やポリペプチドを蛍光標識する試薬キットです。AE-1430 EzApply¥9,800 タンパク質のSDS-PAGE用のサンプル調製用の試薬キットです。e-PAGEL HR ミニサイズ既製ゲル¥18,800
タンパク質のSDS-PAGE用のサンプル調製用の試薬キットです。e-PAGEL HR ミニサイズ既製ゲル¥18,800 標準ミニスラブサイズ(90×83mm)のポリアクリルアミド既製ゲルです。高速泳動に対応した、e-PAGELのハイグレードタイプです。WSE-1150P/M パジェラン Ace¥128,000
標準ミニスラブサイズ(90×83mm)のポリアクリルアミド既製ゲルです。高速泳動に対応した、e-PAGELのハイグレードタイプです。WSE-1150P/M パジェラン Ace¥128,000 標準的なミニスラブサイズのポリアクリルアミドゲル電気泳動(PAGE)用電源一体型電気泳動装置です。 WSE-4125 パワードブロット2M¥218,000
標準的なミニスラブサイズのポリアクリルアミドゲル電気泳動(PAGE)用電源一体型電気泳動装置です。 WSE-4125 パワードブロット2M¥218,000 電源付きのセミドライブロッティング装置です。ミニスラブゲル2枚まで同時にブロッティングできます。WSE-4051 クリアブロット・P+膜_M¥20,800
電源付きのセミドライブロッティング装置です。ミニスラブゲル2枚まで同時にブロッティングできます。WSE-4051 クリアブロット・P+膜_M¥20,800 ミニゲルサイズ(8.5×9cm)にカットされた、ブロッティング用PVDF膜です。低バックグラウンドで高感度に検出できます。WSE-4061 クリアブロット・P膜 (低蛍光)_M¥14,800
ミニゲルサイズ(8.5×9cm)にカットされた、ブロッティング用PVDF膜です。低バックグラウンドで高感度に検出できます。WSE-4061 クリアブロット・P膜 (低蛍光)_M¥14,800 ミニゲルサイズ(8.5×9cm)にカットされた、自家蛍光の低いPVDF膜です。蛍光ウェスタンブロッティングの蛍光検出に最適です。WSE-7160 EzStain AQua MEM¥24,800
ミニゲルサイズ(8.5×9cm)にカットされた、自家蛍光の低いPVDF膜です。蛍光ウェスタンブロッティングの蛍光検出に最適です。WSE-7160 EzStain AQua MEM¥24,800 ブロッティング膜上のタンパク質を検出するCBB染色・脱色キットです。脱色後は抗体反応が可能で、トータルタンパク質によるノーマライズにも使用できます。WSE-6370LuminoGraph Ⅲ Lite¥2,800,000~
ブロッティング膜上のタンパク質を検出するCBB染色・脱色キットです。脱色後は抗体反応が可能で、トータルタンパク質によるノーマライズにも使用できます。WSE-6370LuminoGraph Ⅲ Lite¥2,800,000~ LuminoGraph Ⅲ Liteは、高解像度6メガピクセル冷却CCDカメラと、高感度F0.8レンズを搭載したハイエンド ケミルミ/蛍光 撮影システムです。画像解析ソフトウェア CS Analyzer 4 (Windows版)¥250,000
LuminoGraph Ⅲ Liteは、高解像度6メガピクセル冷却CCDカメラと、高感度F0.8レンズを搭載したハイエンド ケミルミ/蛍光 撮影システムです。画像解析ソフトウェア CS Analyzer 4 (Windows版)¥250,000 ATTO CS Analyzer 4は、発光検出画像やゲル画像などの電気泳動パターンの濃度定量、分子量計測などを行う画像解析ソフトウエアです。
ATTO CS Analyzer 4は、発光検出画像やゲル画像などの電気泳動パターンの濃度定量、分子量計測などを行う画像解析ソフトウエアです。